



 April 18th, 2024
April 18th, 2024
Comparison OF The Key Differences Between The MDR And IVDR In The EU
In the fast-paced world of medical devices, staying updated on regulatory changes is crucial for manufacturers, healthcare professionals, and patients alike. In the European Union (EU), two significant regulations have been introduced to enhance the safety and effectiveness of medical devices – the In Vitro Diagnostic Regulation (IVDR) and the Medical Device Regulation (MDR).
The need for new regulations for medical devices and in vitro medical devices (IVD) arises from the dynamic nature of the healthcare industry and advancements in technology. The regulatory landscape for these devices was recognized as outdated and insufficient to address the evolving challenges and risks associated with their use. The European Union responded to these concerns by introducing the EU Medical Device Regulation (MDR) and In Vitro Diagnostic Device Regulation (IVDR).
The key objectives of the new regulations, Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR), are to enhance the identification of medical devices, standardize data across the industry, and incorporate technological advancements. These regulations strive to establish a robust framework that guarantees the safety, effectiveness, and quality of medical devices.
This blog aims to provide a comprehensive overview of both regulations, highlighting their key points and key differences.

I. IN VITRO DIAGNOSTIC REGULATION (IVDR):
The In Vitro Diagnostic Regulation (IVDR), Regulation (EU) 2017/746, was adopted by the European Parliament and the Council in 2017 and came into effect on 25 May 2017. It replaced the previous In Vitro Diagnostic Directive (IVDD) 98/79/EC and introduced a more stringent framework for the approval and marketing of in vitro diagnostic medical devices (IVDs). This regulation specifically pertains to in vitro diagnostic medical devices distributed within the European Union.
The implementation of In Vitro Diagnostic Regulation (IVDR) introduces a more stringent regulatory framework compared to its predecessor, the IVDD. Companies engaged in the manufacturing of in vitro diagnostic medical devices are subject to the In Vitro Diagnostic Regulation (IVDR), with the deadline for compliance set at May 2022. Compliance involves the submission of comprehensive technical documentation, which is a prerequisite for authorization and market entry.
The transition to In Vitro Diagnostic Regulation (IVDR) signifies a great shift in the regulatory pathway for in vitro diagnostic devices. It places a greater emphasis on the stringent evaluation of technical documentation to meet the new requirements. This shift is a proactive response to the dynamic nature of the industry, aligning regulatory practices with the advancements and complexities observed in diagnostic technologies.
Key Points:
1. Scope and Definition: In Vitro Diagnostic Regulation (IVDR) broadens the scope of IVDs, encompassing a wider range of products used for the examination of specimens derived from the human body. This includes tests for diseases, infections, and genetic conditions.
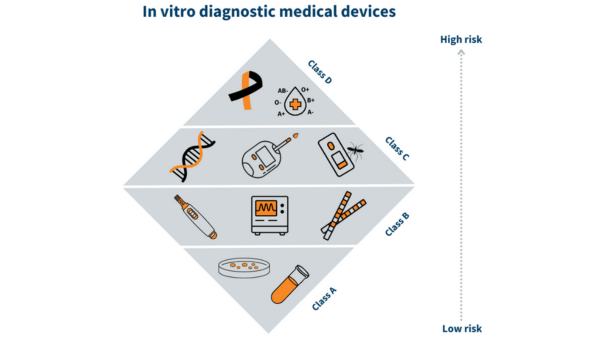
2. Risk Classification: Like Medical Device Regulation (MDR), In Vitro Diagnostic Regulation (IVDR) adopts a risk-based classification system. Devices are categorized into Classes A, B, C, or D based on their inherent risks, with higher classes subject to more stringent regulatory requirements.
3. Conformity Assessment: Manufacturers must undergo a conformity assessment procedure, involving a rigorous evaluation of technical documentation and quality management systems by a notified body.
4. Unique Device Identification (UDI): In Vitro Diagnostic Regulation (IVDR) mandates the use of a UDI system for traceability and post-market surveillance. This ensures the identification and tracking of devices throughout their lifecycle.
II. MEDICAL DEVICE REGULATION (MDR):
The Medical Device Regulation (MDR), Regulation (EU) 2017/745, was also adopted in 2017 and came into force on May 26, 2021. It replaces the Medical Device Directive (MDD) 93/42/EEC and aims to strengthen the regulatory framework for medical devices, ensuring a higher level of safety and efficacy.
The introduction of EU Medical Device Regulation (MDR) is a response to the evolving landscape of the medical device industry, necessitating updates to regulations to ensure alignment with technological advancements and improved patient safety. The primary goals of EU Medical Device Regulation (MDR) include enhancing the identification of medical devices, standardizing data across the industry, and incorporating advancements in medical technology.
The transition from MDD to EU Medical Device Regulation (MDR) is crucial for manufacturers and suppliers of medical devices. MDD-certified devices are required to undergo certification according to the new EU Medical Device Regulation (MDR) requirements by 25 May 2024, or earlier if the MDD certification expires before this date. This transition period allows for a gradual shift towards compliance with the more stringent and contemporary regulations outlined in EU Medical Device Regulation (MDR).
One of the key promises of the EU Medical Device Regulation (MDR) framework is an improvement in transparency, predictability, robustness, and sustainability compared to the previous regulatory framework. Transparency ensures clear and accessible information, predictability allows for better planning and compliance, robustness ensures the regulations are strong and effective, and sustainability ensures that the framework remains relevant in the face of ongoing advancements in the medical field.
In essence, the EU Medical Device Regulation (MDR) signifies a commitment to ensuring that medical devices adhere to high standards of safety and efficacy. The regulation acknowledges the dynamic nature of the medical device industry and seeks to establish a framework that is not only more contemporary but also more transparent and reliable for manufacturers, suppliers, and, most importantly, for the well-being of patients relying on these medical devices.
Key Points:
1. Scope and Definition: Medical Device Regulation (MDR) expands the definition of medical devices, incorporating products not previously regulated. It includes devices intended for aesthetic purposes and those with non-medical purposes but with a medical intended purpose.
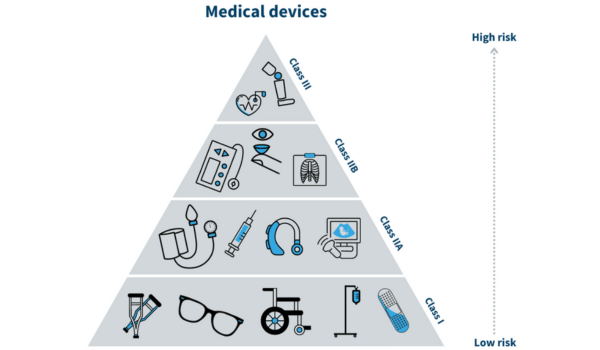
2. Risk Classification: Similar to In Vitro Diagnostic Regulation (IVDR), Medical Device Regulation (MDR) classifies devices based on risk. The classification ranges from Class I (lowest risk) to Class III (highest risk), with corresponding conformity assessment requirements.
3. Conformity Assessment: Manufacturers must undergo a conformity assessment procedure, with higher-risk devices requiring the involvement of a notified body. The assessment considers clinical data, risk management, and post-market surveillance.
4. UDI system: Medical Device Regulation (MDR) introduces a UDI system for medical devices, enabling better traceability and improving post-market surveillance.
THE KEY DIFFERENCES BETWEEN In Vitro Diagnostic Regulation (IVDR) AND Medical Device Regulation (MDR):
In scientific terms, the EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) differ in their scope, pre-market data requirements, and post-market obligations. The highlighting differences between the medical device regulations and In Vitro Diagnostics Medical Device Regulations are as follows:
TERMSEU MDREU IVDRREGULATIONREGULATION (EU) 2017/745REGULATION (EU) 2017/746APPLICABILITYMedical Devices for human use manufactured or sold into the European UnionIn vitro diagnostic medical devices for human use manufactured or sold into the European Union.ARTICLES123 articles, applied to all human medical devices113 articles, focuses solely on in vitro diagnostic devicesPRE-MARKET DATAClinical Evaluation report based on evaluation of clinical evidence or a clinical investigationPerformance evaluation and performance studies to justify intended patient outcomeRISK CLASSIFICATIONClass I (Low risk)
- Class Is (Sterile)
- Class Im (Measurable)
- Class Ir (reusable)
Class IIa (Low to Medium risk)
Class IIb (Medium to High risk)
Class III (High risk)
Class A (Low risk)
Class B (Medium)
Class C (Medium to High)
Class D (High)
POST-MARKET DATAPost-Market Clinical Follow-UpPost-Market Performance (Surveillance and Vigilance)NOTIFIED BODIESApplicable to all Class IIa, IIb and III devicesApplicable to all Class B, C and D devicesCLINICAL EVIDENCEFocused mainly on safety and clinical performance in accordance to the intended purpose of the deviceFocused mainly on performance of device and improved patient outcomesPOST MARKET SURVEILLANCE REPORT (PMSR)Required for Class I manufacturers – updated when necessary)Required for Class A and Class B manufacturers – updated when necessary)PERIODIC SAFETY UPDATE REPORT (PSUR)Required for Class IIa, IIb and III manufacturers – updated at least annuallyRequired for Class C and D manufacturers – updated at least annuallyGENERAL SAFETY AND PERFORMANCE REQUIREMENTS23 applicable clauses as per Regulation (EU) 2017/74520 applicable clauses as per Regulation (EU) 2017/746
CONCLUSION:
Navigating the regulatory landscape of medical devices in the European Union demands a thorough understanding of both the In Vitro Diagnostic Regulation (IVDR) and the Medical Device Regulation (MDR). While these regulations share commonalities, such as risk-based classification and conformity assessment, differences in scope, implementation dates, and the level of notified body involvement necessitate careful consideration from manufacturers. Adhering to these regulations is not only a legal requirement but a commitment to ensuring the safety and efficacy of medical devices for the well-being of patients.

REFERENCES:
1. REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC
2. REGULATION (EU) 2017/746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU
Comments